About Capillary Electrophoresis
Sanger sequencing is a method of DNA sequencing first commercialized by Applied Biosystems, based on the selective incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication.
Developed by Frederick Sanger and colleagues in 1977, it was the most widely used sequencing method for approximately 40 years. More recently, higher volume Sanger sequencing has been supplanted by "Next-Gen" sequencing methods, especially for large-scale, automated genome analyses.
However, the Sanger method remains in wide use, for smaller-scale projects, validation of Next-Gen results and for obtaining especially long contiguous DNA sequence reads (> 500 nucleotides).
About the Method
The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleosidetriphosphates (dNTPs), and modified di-deoxynucleotidetriphosphates (ddNTPs), the latter of which terminate DNA strand elongation. These chain-terminating nucleotides lack a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a modified ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labelled for detection in automated sequencing machines.
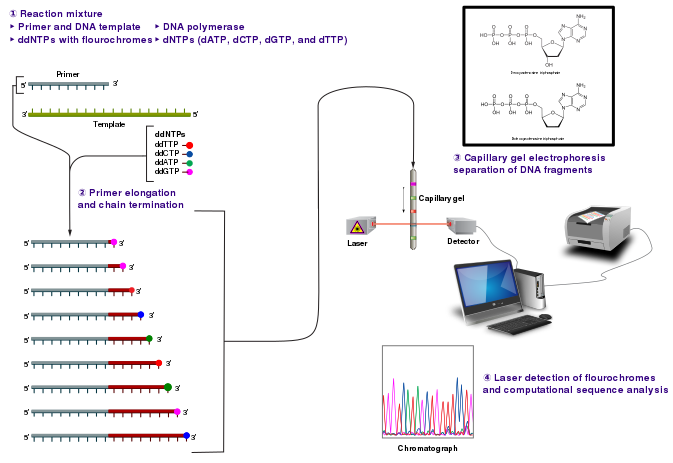
The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP), while the other added nucleotides are ordinary ones. The dideoxynucleotide concentration should be approximately 100-fold lower than that of the corresponding deoxynucleotide (e.g. 0.005mM ddTTP : 0.5mM dTTP) to allow enough fragments to be produced while still transcribing the complete sequence.
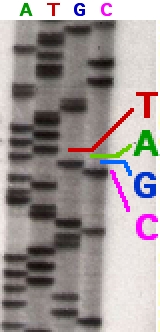
Following rounds of template DNA extension from the bound primer, the resulting DNA fragments are heat denatured and separated by size using gel electrophoresis. The DNA bands may then be visualized by autoradiography or UV light and the DNA sequence can be directly read off the X-ray film or gel image.
In the image on the right, X-ray film was exposed to the gel, and the dark bands correspond to DNA fragments of different lengths. A dark band in a lane indicates a DNA fragment that is the result of chain termination after incorporation of a dideoxynucleotide (ddATP, ddGTP, ddCTP, or ddTTP). The relative positions of the different bands among the four lanes, from bottom to top, are then used to read the DNA sequence.
Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5' end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis and automation. The later development by Leroy Hood and coworkers of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing.
Chain-termination methods have greatly simplified DNA sequencing. For example, chain-termination-based kits are commercially available that contain the reagents needed for sequencing, pre-aliquoted and ready to use. Limitations include non-specific binding of the primer to the DNA, affecting accurate read-out of the DNA sequence, and DNA secondary structures affecting the fidelity of the sequence.
Dye-terminator sequencing utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction, rather than four reactions as in the labelled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, each of which emit light at different wavelengths. Owing to its greater expediency and speed, dye-terminator sequencing is now the mainstay in automated sequencing.
Automation and sample preparation
Automated DNA-sequencing instruments (DNA sequencers) can sequence up to 384 DNA samples in a single batch. Batch runs may occur up to 24 times a day. DNA sequencers separate strands by size (or length) using capillary electrophoresis, they detect and record dye fluorescence, and output data as fluorescent peak trace chromatograms. Sequencing reactions (thermocycling and labelling), cleanup and re-suspension of samples in a buffer solution are performed separately, before loading samples onto the sequencer. A number of commercial and non-commercial software packages can trim low-quality DNA traces automatically. These programs score the quality of each peak and remove low-quality base peaks (which are generally located at the ends of the sequence). The accuracy of such algorithms is inferior to visual examination by a human operator, but is adequate for automated processing of large sequence data sets.
Microfluidic Sanger sequencing
Microfluidic Sanger sequencing is a lab-on-a-chip application for DNA sequencing, in which the Sanger sequencing steps (thermal cycling, sample purification, and capillary electrophoresis) are integrated on a wafer-scale chip using nanoliter-scale sample volumes. This technology generates long and accurate sequence reads, while obviating many of the significant shortcomings of the conventional Sanger method (e.g. high consumption of expensive reagents, reliance on expensive equipment, personnel-intensive manipulations, etc.) by integrating and automating the Sanger sequencing steps.
In its modern inception, high-throughput genome sequencing involves fragmenting the genome into small single-stranded pieces, followed by amplification of the fragments by Polymerase Chain Reaction (PCR). Adopting the Sanger method, each DNA fragment is irreversibly terminated with the incorporation of a fluorescently labeled dideoxy chain-terminating nucleotide, thereby producing a DNA “ladder” of fragments that each differ in length by one base and bear a base-specific fluorescent label at the terminal base. Amplified base ladders are then separated by Capillary Array Electrophoresis (CAE) with automated, in situ “finish-line” detection of the fluorescently labeled ssDNA fragments, which provides an ordered sequence of the fragments. These sequence reads are then computer assembled into overlapping or contiguous sequences (termed "contigs") which resemble the full genomic sequence once fully assembled.
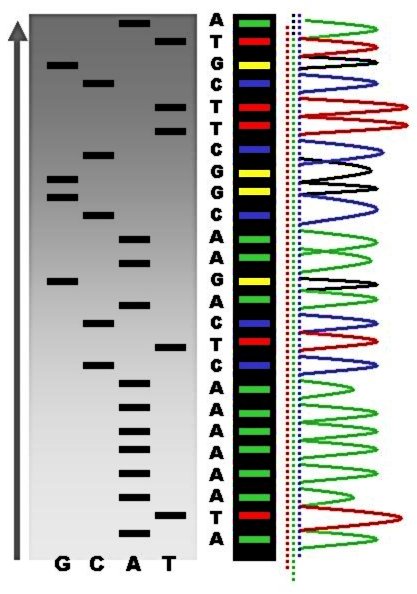
Sequence ladder by radioactive sequencing compared to fluorescent peaks
Sanger methods achieve read lengths of approximately 800bp (typically 500-600bp with non-enriched DNA). The longer read lengths in Sanger methods display significant advantages over other sequencing methods especially in terms of sequencing repetitive regions of the genome. A challenge of short-read sequence data is particularly an issue in sequencing new genomes (de novo) and in sequencing highly rearranged genome segments, typically those seen of cancer genomes or in regions of chromosomes that exhibit structural variation.
Other useful applications of DNA sequencing include single nucleotide polymorphism (SNP) detection, single-strand conformation polymorphism (SSCP) heteroduplex analysis, and short tandem repeat (STR) analysis. Resolving DNA fragments according to differences in size and/or conformation is the most critical step in studying these features of the genome.
Sequencing chemistry:
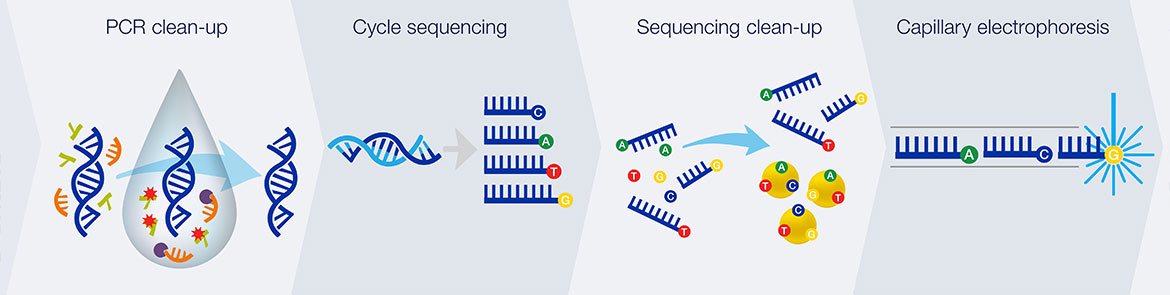
1. Thermal cycling
In the TC reaction chamber, dye-terminator sequencing reagent, template DNA, and primers are loaded into the TC chamber and thermal-cycled for 35 cycles ( at 95 °C for 12 seconds and at 60 °C for 55 seconds).
2. Purification
The charged reaction mixture (containing extension fragments, template DNA, and excess sequencing reagent) is conducted through a capture/purification chamber at 30 °C via a 33-Volts/cm electric field applied between capture outlet and inlet ports. The capture gel through which the sample is driven, consists of 40 μM of oligonucleotide (complementary to the primers) covalently bound to a polyacrylamide matrix. Extension fragments are immobilized by the gel matrix, and excess primer, template, free nucleotides, and salts are eluted through the capture waste port. The capture gel is heated to 67-75 °C to release extension fragments.
3. Capillary electrophoresis
Extension fragments are injected into the CE chamber where they are electrophoresed through a 125-167-V/cm field.
|
Required materials and reagents for SeqStudio Genetic Analyser
|
|
|
P/N |
Description |
|
A33671 |
CARTRIDGE ASSAY |
|
A33401 |
SEQSTUDIO CATHODE BUFFER CONTAINER |
|
A35640 |
CE RESERVOIR SEPTA PIECES |
|
A35641 |
96-well Septa for SeqStudio™ Genetic Analyzer |
|
A35641 |
CE PLATE 96-WELL PIECES |
|
4337455 |
BDT V3.1 RR-100 & SEQ BUFFER |
|
4336697 |
TUBE-5X SEQ BUFFER SMALL 1mL |
|
4311320 |
HI-DI FORMAMIDE BOTTLE |
|
4376486 |
BIGDYE XTERMINATOR KIT 2ML |
|
Required materials and reagents for 3500 Genetic Analyser
|
|
|
P/N |
Description |
|
4404685 |
CAPILLARY ARRAY 8-CAP 50CM RUO |
|
4393708 |
POP-7 (384)POLYMER 3500 SERIES |
|
4393927 |
ANODE BFFR CONTAINR 3500SERIES |
|
4408256 |
CATHODE BFR CONTAINR 3500 SER |
|
4393718 |
CONDITIONING REAGNT 3500SERIES |
|
4410715 |
Septa Cathode Buffer Container (for the 3500 series Genetic analyzers) |
|
4412614 |
Septa for Genetic Analyzers, 96 well |
|
A35641 |
CE PLATE 96-WELL PIECES |
|
4337455 |
BDT V3.1 RR-100 & SEQ BUFFER |
|
4336697 |
TUBE-5X SEQ BUFFER SMALL 1mL |
|
4311320 |
HI-DI FORMAMIDE BOTTLE |
|
4376486 |
BIGDYE XTERMINATOR KIT 2ML |